Input of molecular NMR parameters via a YAML file
NMR parameters for molecules can be provided in a YAML file. A brief summary of relevant YAML features is provided before proceeding to more detailed explanations and examples.
- Dictionaries in YAML are defined as
key: valuepairs. Most commonly, a dictionary contains one key/value pair per line:key 1: value 1 key 2: value 2 value 1is interpreted as a string,1is interpreted as an integer and1.0is interpreted as a floating-point number. To avoid problems with special characters (e.g., square brackets), strings may be enclosed in single or double quotes (they have different meanings, and single quotes should be preferred for a literal interpretation of the string).- Lists can be defined over multiple lines as:
Lists can also be enclosed in square brackets:- item 1 - item 2[item 1, item 2, item 3]. The nested list[[1, 2, 3], [4, 5, 6]]is equivalent to:- [1, 2, 3] - [4, 5, 6] - Indentation is part of the syntax: key/value pairs or list entries over multiple lines need to have the same number of leading spaces (no tabs).
- Comments start with a hash,
#.
Definition of the molecular structure
Definition using SMILES
A molecular structure needs to be provided along with its NMR parameters in order to get a complete molecular data input. The YAML input accepts 2D structural representations, i.e., SMILES strings or Molfiles.
The simplest way to define a structure in the input file is through its SMILES representation. This is done using the key smiles, followed by a representation of the molecule. For acetic acid:
# Acetic acid defined using SMILES.
smiles: CC(=O)O
SMILES strings often contain square brackets [...]. In such cases, the string should be enclosed within quotes, '...', to avoid problems with the YAML parser.
Definition using a Molfile
Manual definition of increasingly large molecules using SMILES can be cumbersome. For example, the string representation of penicillin V would be:
smiles: 'CC1(C)S[C@@H]2[C@H](NC(=O)COc3ccccc3)C(=O)N2[C@H]1C(=O)O'
Instead, it is easier to draw a graphical representation such as the one below using one of many available proprietary or open source packages.
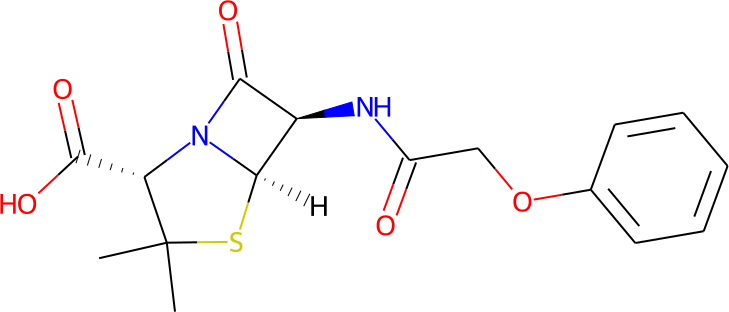
Such structural 2D representations are commonly stored in Molfiles. A molecule can be read from a Molfile by specifying the file name after the key molfile:
molfile: penicillin_v.mol
Note that the YAML input needs to contain either a Molfile or SMILES, but it is not possible to specify both at the same time. Both the V2000 and V3000 variants of the Molfile specification are supported in the input.
Hydrogens in the molecular structure
2D representations of molecular structures, whether as skeletal formulas or as SMILES, tend to omit hydrogens. Instead, the number of hydrogen atoms is inferred from the atomic valencies, especially those of the carbons. Any of the following three structures can be provided as a Molfile for the acrylamide molecule:
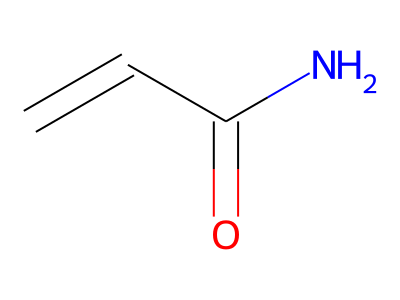
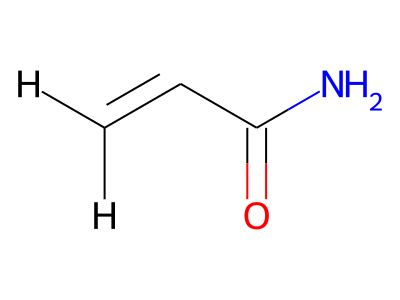
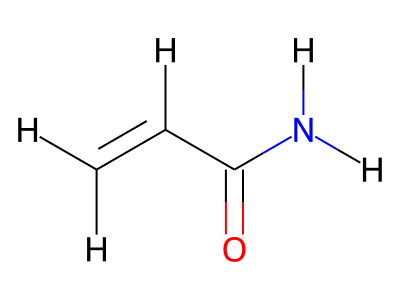
Where hydrogens are suppressed (not drawn out as separate atoms with a bond), their NMR parameters are specified through assignment to the respective skeletal atom. In the leftmost of the three structures shown above, it would be not possible to assign different parameters to the two protons in the CH2 group. Instead, one of the two other structures shown above could be used to specify different parameters for those protons.
The only restriction with regards to hydrogens is that any skeletal atom can be connected either to suppressed or to stand-alone hydrogens, but not to a mixture of both. Thus, the following two structures would be rejected during input parsing:
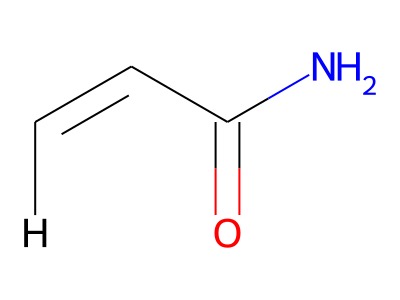
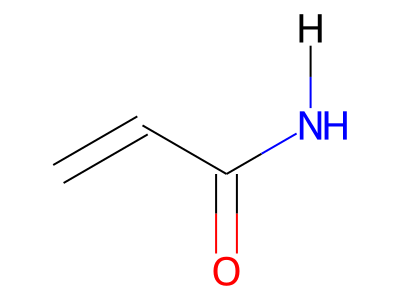
The structure to the left mixes a non-suppressed hydrogen with a suppressed "implicit" hydrogen (CH) on the carbon atom; the structure to the right mixes a non-suppressed hydrogen with a suppressed "explicit" hydrogen (NH) on the nitrogen atom.
Numbering of atoms
Atoms in the structural representation of the molecule are labelled with integers for the assignment of parameters. Indices can be counted starting from zero or from one. To avoid errors or misunderstandings, it is mandatory to specify a count from key in the input file, followed by either 0 or 1. The choice between those two options is entirely arbitrary.
An example for acetamide with atom counting starting from zero:
# Atom indices are 0, 1, 2, 3 in their order of appearance in the SMILES string.
smiles: CC(=O)N
count from: 0
An example for atom counting starting from one:
# Atom indices are 1, 2, 3, 4 in their order of appearance in the SMILES string.
smiles: CC(=O)N
count from: 1
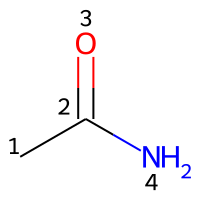
The atoms in a molecule are indexed by their order of appearance in the Molfile. Acetamide may be represented by a Molfile with the following content:
RDKit 2D
4 3 0 0 0 0 0 0 0 0999 V2000
0.0000 0.0000 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
1.2990 0.7500 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
1.2990 2.2500 0.0000 O 0 0 0 0 0 0 0 0 0 0 0 0
2.5981 -0.0000 0.0000 N 0 0 0 0 0 0 0 0 0 0 0 0
1 2 1 0
2 3 2 0
2 4 1 0
M END
Counting the atoms starting from 1 (count from: 1), the appropriate indices are represented in the image below.
Chemical shifts
Providing chemical shifts will be illustrated using the nitrobenzene molecule as an example. Its molecular structure is contained in the file PhNO2.mol and shown in the picture below, including a numbering of its atoms.
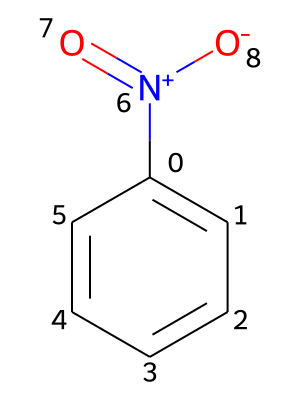
All chemical shifts are specified under the key shifts. Additionally, the values need to be nested under keys representing isotopes, which contain the atomic mass number and the element symbol (e.g., 1H or 13C). For each isotope, the chemical shifts are provided in pairs of an atomic index and the associated value in ppm (parts per million):
# Structure with suppressed protons.
molfile: PhNO2.mol
count from: 0
shifts:
# chemical shifts for protons in ppm
1H:
1: 8.23
2: 7.56
3: 7.71
4: 7.56
5: 8.23
# chemical shifts for carbon-13 nuclei in ppm
13C:
0: 148.5
1: 123.7
2: 129.4
3: 134.3
4: 129.4
5: 123.7
To assign chemical shifts for suppressed protons (that are not provided explicitly in the skeletal structure), the indices of the respective non-hydrogen atoms are used instead. All suppressed protons connected to the same atom are assigned an identical shift value.
If a Molfile contains hydrogens as standalone atoms, the chemical shifts are assigned to those protons using their respective atom indices. This is illustrated using the file PhNO2_allH.mol. Its structure is shown below.
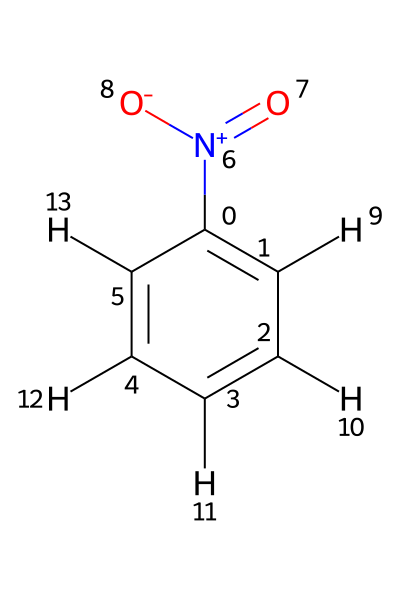
In this example, the 1H shifts need to be assigned to atoms 9-13. Assigning them to atoms 1-5, as in the previous example, would produce an error.
# Structure with suppressed protons.
molfile: PhNO2_allH.mol
count from: 0
shifts:
# chemical shifts for protons in ppm
1H:
9: 8.23
10: 7.56
11: 7.71
12: 7.56
13: 8.23
# chemical shifts for carbon-13 nuclei in ppm
13C:
0: 148.5
1: 123.7
2: 129.4
3: 134.3
4: 129.4
5: 123.7
Indirect spin-spin coupling constants
Indirect spin-spin coupling constants are provided under the key J-couplings in the YAML file. Additionally, the coupling constant values need to be grouped together by isotopes. Keys for each combination of isotopes are combined as isotope1-isotope2: e.g., 1H-1H for coupling constants between two protons or 1H-13C for the associated heteronuclear coupling.
J-coupling constants in units of Hz for each combination of nuclei are provided as a list of lists with the following structure:
J-couplings:
isotope1-isotope2:
- [atom index 1, atom index 2, coupling constant in Hz]
- [atom index 1, atom index 2, coupling constant in Hz]
- [...]
isotope1-isotope2:
- [atom index 1, atom index 2, coupling constant in Hz]
- [...]
The first atom index refers to the first isotope and the second atom index refers to the second isotope. As with shifts, values for suppressed hydrogens are assigned via the associated skeletal carbon or heteroatom. If multiple protons are connected to the same skeletal atom, they are assigned the same coupling constant. Inequivalent protons attached to the same skeletal atom, need to be specified explicitly as standalone atoms in the molecule definition, so that they can be referred to via their respective atom indices.
Examples
1H parameters for propane with SMILES input
smiles: CCC
count from: 0
shifts:
1H:
0: 0.9
1: 1.3
2: 0.9
J-couplings:
1H-1H:
- [0, 1, 7.26]
- [1, 2, 7.26]
The protons at the terminal CH3 groups are assigned chemical shifts of 0.9 ppm each, and the protons of the central CH2 group are given a value of 1.3 ppm. J-couplings between all protons of the neighboring CH3 and CH2 groups are assigned as 7.26 Hz. While an indirect spin-spin coupling interaction exists between equivalent protons within the CH3 and CH2 groups, it is not observed in the spectrum and the associated values are left out.
1H parameters for acrylonitrile
The structure of acrylonitrile is provided as a Molfile in acrylonitrile.mol. Its depiction is shown below.
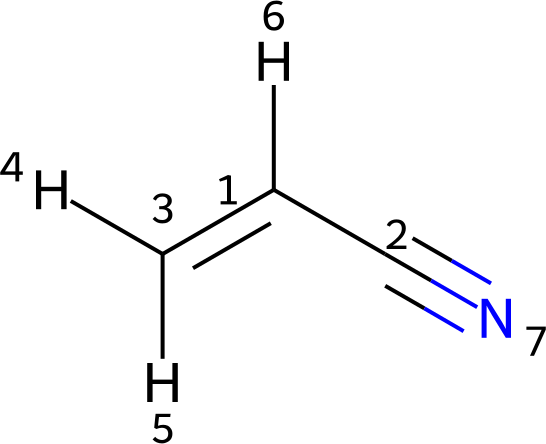
Since protons 4 and 5 are inequivalent, they are specified as standalone atoms with different parameters. In addition, hydrogen 6 is represented as a standalone atom, though suppressing it would also be an equally valid choice.
molfile: acrylonitrile.mol
count from: 1
shifts:
1H:
4: 5.79 # H(trans)
5: 5.97 # H(cis)
6: 5.48 # H(gem)
J-couplings:
1H-1H:
- [4, 5, 0.9]
- [4, 6, 11.8]
- [5, 6, 17.9]
Combined 1H and 13C parameters for chloromethane
To illustrate the definition of heteronuclear coupling constants, the following example shows parameters for 13C-enriched chloromethane. The parameters include the shifts of the three protons and the 13C nucleus, as well as the coupling constants between these nuclei.
# CH3Cl with 13C, the hydrogens inside square brackets are implicit.
smiles: '[13CH3]Cl'
# C will have index 1 and Cl will have index 2
count from: 1
shifts:
# Shifts of the three protons.
1H:
1: 3.05
# Shift of carbon-13 in the molecule.
13C:
1: 25.6
J-couplings:
# Coupling between the three protons (would normally not be observed).
1H-1H:
- [1, 1, -10.8]
# Coupling between the three protons and the 13C atom.
1H-13C:
- [1, 1, 150.0]
Stored YAML files
For the molecules included in the examples module of the hqs_nmr_parameters package, YAML input files (and Molfiles when indicated as molecular structure) are available in the file system. To access them, use the following command, taking into account that each YAML file takes the name of its key in the data set. For acrylonitrile (with key "C2H3CN"), we will have:
from pathlib import Path
from hqs_nmr_parameters import examples
identifier = "C2H3CN"
acrylonitrile_yaml = Path(examples.__file__).parent.joinpath("parameters", identifier + ".yaml")
print(acrylonitrile_yaml.read_text())
name: Acrylonitrile
molfile: C2H3CN.mol
count from: 1
shifts:
1H:
4: 5.79 # H(trans)
5: 5.97 # H(cis)
6: 5.48 # H(gem)
J-couplings:
1H-1H:
- [4, 5, 0.9]
- [4, 6, 11.8]
- [5, 6, 17.9]
description: |
1H parameters for acrylonitrile.
Values were obtained from Hans Reich's Collection, NMR Spectroscopy.
https://organicchemistrydata.org
As we have seen, to get this data as MolecularData instance:
from hqs_nmr_parameters.examples import molecules
identifier = "C2H3CN"
parameters = molecules[identifier]
print(parameters.shifts)
[(3, 5.79), (4, 5.97), (5, 5.48)]
Therefore, even if the atom counting in the YAML file starts at 1, the numbering in MolecularData always starts at 0.
Reading and processing YAML input files
In the HQS NMR Tool, a YAML file containing NMR parameters is parsed using the read_parameters_yaml function
from the hqs_nmr_parameters package:
from hqs_nmr_parameters import read_parameters_yaml
parameters = read_parameters_yaml("input_file.yaml")
The read_parameters_yaml function can read the following keywords from a YAML file:
name: An optional name of the molecule.shifts: Chemical shifts in format {isotope: {index: value}}.j_couplings/J-couplings: J-coupling values in format {isotope1-isotope2: [[index1, index2, value], ...]}.count from/count_from: Specifies whether to count atomic indices starting from zero or from one.smiles: SMILES string of the molecule (better enclosed in quotation marks).molfile: Path to a Molfile with the molecular structure.molblock: Compressed Molfile content (not in clear text).temperature: Temperature in K.solvent: Name of the solvent.description: Additional further description.
parameters is an instance of the Pydantic MolecularData class.