Introduction
HQS Molecules is a user-friendly Python package designed to streamline computational chemistry workflows, with a focus on low-cost stages. Looking up a molecule name on PubChem, converting its SMILES string to an XYZ structure, optimizing its geometry and performing a conformer search: with HQS Molecules, all these steps take just a few lines of Python code. Wrappers around RDKit, Open Babel, xTB and CREST bundle these diverse open-source packages behind a uniform interface that is compliant with their respective licenses, while improving upon critical features. This makes HQS Molecules valuable both as a standalone tool for computational chemists, as well as a component in conjunction with other HQStage modules by HQS Quantum Simulations GmbH.
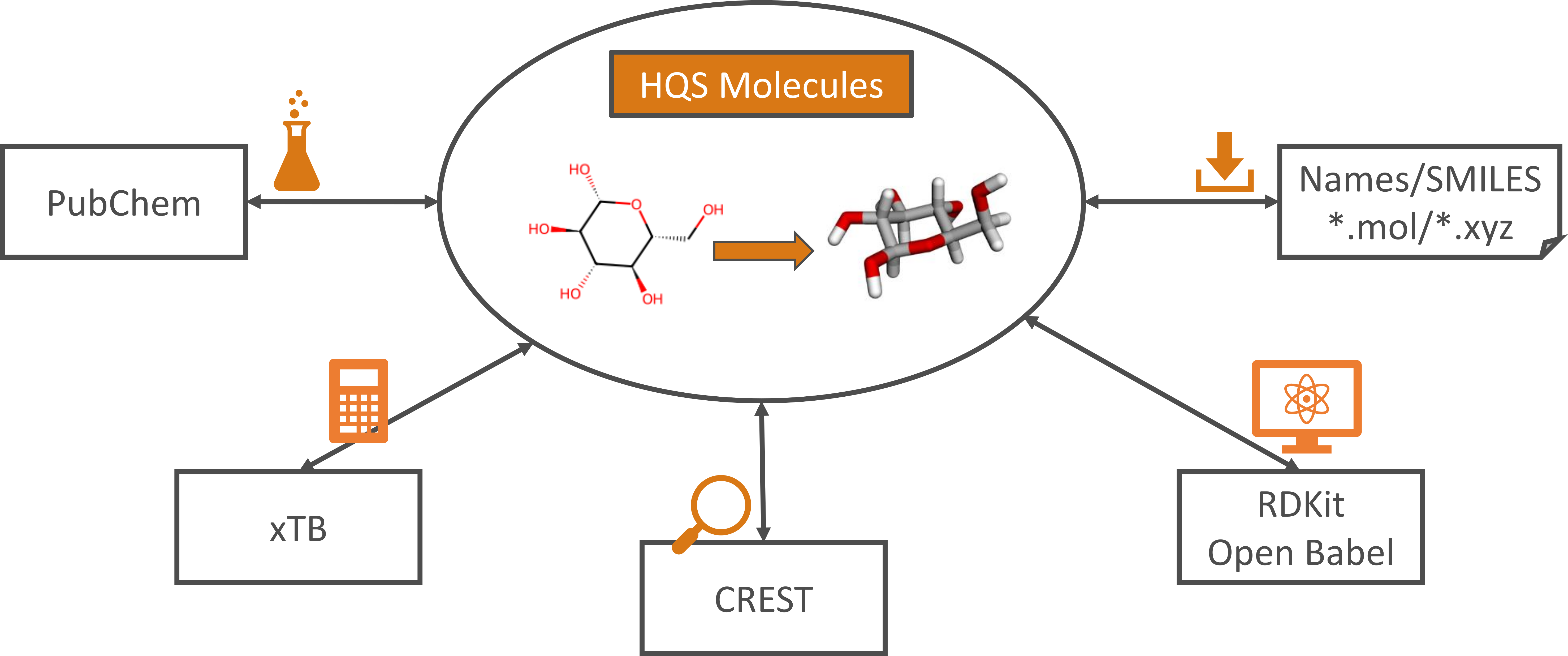
Features
HQS Molecules provides the following functionality to its users:
- Converting chemical structures from 2D to 3D representations. The RDKit and Open Babel packages are used to create XYZ structures from SMILES strings
or Molfiles, with an additional check for the correctness of the generated geometry. Standard chemistry literature and databases commonly encode molecules as representations of their 2D structural formula. On the other hand, 3D geometries are required to perform quantum-chemical calculations.
HQS Moleculesallows chemists and researchers in other fields to generate these conveniently and automatically. - API access to the PubChem database for name-to-structure and structure-to-name search. Direct access to the PubChem database via the Python API of
HQS Moleculesenables users to quickly retrieve a chemical structure by providing a compound name, or to search in reverse. This streamlines the process of gathering data for research and development, permitting automatization and reducing the potential for errors in manual searches.HQS Moleculescomplies with PubChem network access quotas and verifies that retrieved data is in the public domain. - Performing single-point energy calculations, geometry optimizations and frequency calculations with the semi-empirical xTB program.
HQS Moleculespermits users to integrate xTB functionality seamlessly into automated and efficient computational chemistry workflows with Python, enhancing productivity and enabling more complex simulations. - Performing conformer search through a Python interface for the CREST program with post-processing. Conformer search is essential for identifying the most stable molecular conformations and for understanding the conformational flexibility of molecules. By providing a Python interface,
HQS Moleculesmakes conformer search more accessible and easier to integrate into computational workflows. An additional refinement procedure for conformer ensembles has been developed exclusively withinHQS Molecules. Robustness and reliability is improved by reassigning conformers and rotamers, by carrying out a completion of the structural ensemble and by screening out transition states or other saddle points. - Reading and writing XYZ files. Being able to handle files with a single or with multiple geometries ensures compatibility with a wide range of computational chemistry tools and facilitates the exchange of molecular data between different software packages.
- Extensive usage of Pydantic to define input and output classes. Pydantic provides extensive functionality for data validation and serialization using Python type annotations. Besides ensuring correctness of data types, this permits input and output to be converted to and from JSON straightforwardly.
Example Use Cases
Generating three-dimensional structures, optimizing geometries, calculating molecular energies or vibration frequencies and sampling molecular conformations are crucial building blocks for molecular simulation workflows in the chemical pharmaceutical industries:
- Quantum chemistry calculations, with density functional theory (DFT) or with other methods, require three-dimensional geometries as their input. This information is rarely present in data sets that do not originate from a computational chemistry context.
HQSused features as implemented inHQS Moleculesto calculate thermodynamic properties of several thousand molecules for a customer with DFT: the starting point was a text file containing little more relevant information than compound names. - Whether you want to model chemical reactions or to calculate spectroscopic parameters:
HQS Moleculescan be used as part of your own Python-based workflow to provide the input for your favorite quantum chemistry software. Build upon the established expertise behind the open-source packages xTB, CREST, RDKit and Open Babel without needing to write your own interface. - Quantum computing algorithms for molecular electronic structure problems usually require a three-dimensional structure as an input, most often in XYZ format. Obtaining or creating this file can be a nuisance for specialists and non-specialists alike. With
HQS Molecules, a molecule name is all you need to generate three-dimensional geometries for a vast number of catalogued compounds. - The Active Space Finder is an open-source tool by
HQS Quantum Simulationsthat builds upon the PySCF software. It provides a user-friendly way to select active spaces for conventional multi-reference calculations and for quantum computing algorithms alike. UseHQS Moleculesto create input geometries for PySCF and the Active Space Finder.
Further Material
This manual contains extensive explanations on the usage of this module. In addition, please check the example notebooks for practical demonstrations of the utilities provided.